
 RAD-seq简介
RAD-seq简介
# RAD-seq 简介:
RADseq (Restriction-site associated DNA sequencing)是在第二代测序基础上发展而来的一项基于全基因组酶切位点的简化的基因组测序技术,是简化基因组测序技术的总称。RADseq不仅实验操作简单,性价比高,更为重要的是它并不依赖参考基因组的信息,一次测序即可获得数以万计的多态性遗传标记,已然广泛用于生态学、遗传学、基因组学等研究领域。
# RAD-seq 技术
RADseq技术是对特定的酶切片段进行高通量测序,根据使用的限制性内切酶的种类和数量,可以将RADseq分为Original RADseq,2b-RADseq,ddRAD, ezRAD, GBS等技术方法。由于采用pooling建库的方式,与Paired-end和Mate-pair文库相比,RADseq技术一次可以构建多至96个测序文库,实验操作相当便利。
不同的RADseq技术所获得的loci的数量差异较大,总体来说:相比于GBS技术,Original RADseq和2b-RADseq可以获得更多的loci,因此,普遍认为这两种方法更适合用于探讨种群的进化关系,种群结构,gene flow等相关问题。
# RAD-seq 在生物进化研究中的应用:
- 生物的适应性: 基于RADseq数据,利用GWAS和Fst outlier两种分析方法发现了与蝴蝶翅膀模式相关的loci,揭示了蝴蝶对生长环境的适应机制。
- 种群进化历史: 三刺鱼不同种群的有效种群大小(Ne),并结合最小等位基因频谱,讨论9个地理种群可能经历的种群瓶颈事件。
- 群体结构: 利用RAD-Tags的分析方法对蚊子的群体结构进行了探讨,发现基于SNP分型数据构建的系统谱系树与蚊子的生长环境基本对应,出现细微偏差的原因很可能在于蚊子生长环境的不同以及人为活动的干扰。
# RADseq中的错误与偏差
等位基因丢失和无效等位基因:当多态性位点正好位于内切酶的酶切位点时,就会造成等位基因丢失;当等位基因位于的片段缺乏完整的酶切位点将不会被测序,成为无效等位基因,会造成基因分型错误。
PCR 重复和分型错误:随机的PCR扩增过程会造成某一个等位基因比例的不均匀性,这种情况下:杂合子很可能会误以为是纯合子。
位点覆盖深度的差异:一般避免出现覆盖度差异的方法是增加单个样本的测序量,这样会导致测序成本的增加。
# 怎么设计一个RADseq研究方案
设计一个RADseq实验方案需要考虑以下几点:
RADseq技术的选择:不同的RADseq的技术的特点各不相同,例如:虽然大部分的RAD可以获得更多的loci,但这些RADseq的文库构建费时费力,并且有些RAD技术对于基因组较为复杂的物种而言,适用性较差,例如2b-RAD。
Reads的长度:Long reads或者Pair-end reads在RADseq技术中拥有更多的优势,包括locus的识别度,旁系同源的污染排除,重复序列等等。尤其针对那些没有参考基因组且基因组重复序列比例较高的物种而言,Long reads和Pair-end reads优势更为明显。
参考基因组信息:虽然RADseq并不完全依赖参考基因组,但拼接较好的基因组对RADseq技术来说是非常有利的。
Coverage:利用reference Mapping的方法进行的RADseq分析,在基因组完整程度较好的情况下,测序覆盖度的要求较低(<1x);而利用de novo分析则需要更深的测序覆盖度(10-20x)。
# 不同RAD-seq技术比较
RRL(Reduced-RepresentationLibraries):简化文库,是最早的简化基因组技术,2000年由Altshuler, D.提出,最初应用于人类样品,后逐渐应用于动植物样品,由于其没有引入barcode ,不能区分混合样品,后续使用较少。RAD及GBS可以说是RRL的升级版。
RAD(Restriction-site Associated DNA):是与限制性核酸内切酶识别位点相关的DNA。2007年由miller提出。
GBS(Genotyping-by-Sequencing):是指通过测序进行基因分型。2011年由Elshire, R. J.提出。
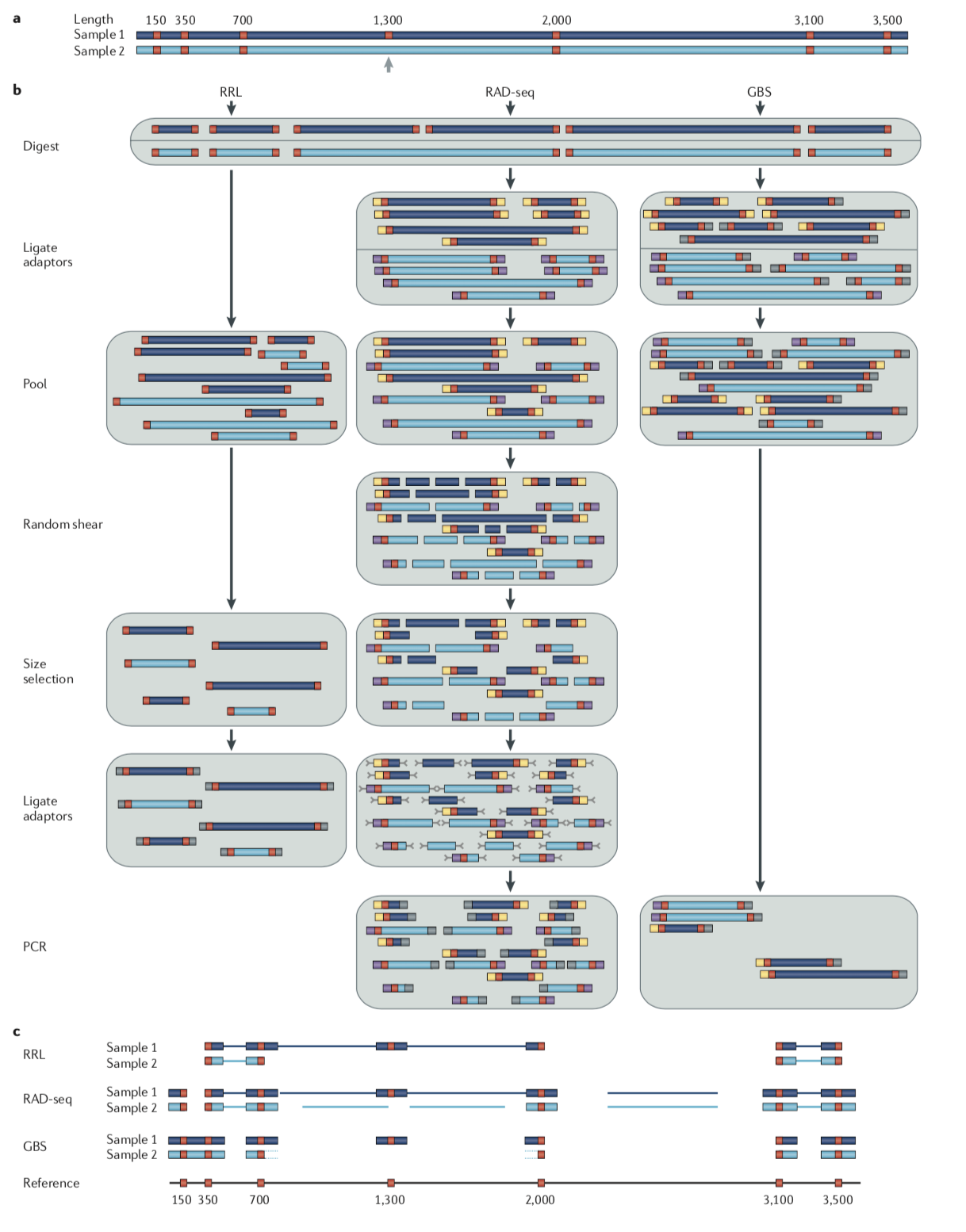
从上图的技术流程对比可以看出:
RRL的优点是步骤少,缺点是没有对不同样品加上不同的barcode,导致后续无法区分单个样品;
RAD-seq的技术优点是不同样品加上特有的barcode,且能把绝大部分酶切位点两侧的片段收集下来,缺点是增加了随机打断、片段选择、加Y型接头等步骤,稍微复杂些;
GBS 优点是不同样品加上的特有的barcode步骤简单,缺点是只能收集短的酶切片段,酶切片段偏少。
近几年简化基因组技术也在不断发展并逐渐分化出不同的分支,如RAD技术就在原来的sd-RAD(single digest-RAD,即常见的单酶切RAD)的基础上改进分化出dd-RAD、2b-RAD; GBS也在原来的sd-GBS(single digest-GBS,即单酶切GBS)的基础上分化出dd-GBS。
dd-RAD(double digest-RAD),即双酶切RAD,2012年由Brant K.提出。采用两种酶对基因组DNA进行酶切,第二种酶的酶切取代了随机打断的作用,优点是不同样品在同一酶切位点处收集的片段长度一致,缺点是获得的片段比单酶切RAD少。
2b-RAD(IIB-RAD),采用IIB型限制性内切酶进行酶切,来产生一致性片段进行测序,由王师教授2012年提出。IIB型限制性内切酶能在识别位点的上游和下游位点分别切段,获得固定长度的片段。优点是片段长度一致,缺点是片段长度较短,一般在33或36bp.
dd-GBS(doubledigest-GBS),即双酶切GBS,采用两种酶对基因组进行酶切,步骤与双酶切RAD非常相似,区别在于pooling的样品数量有所不同。
其实,每一种技术都有自己独特的应用领域,并不能简单的说优点和缺点,比如说GBS采用甲基化敏感酶时虽然收集的片段偏少,但是可以避开高重复区域,收集到的酶切片段及得到的标记有效性更高,比较适合高重复物种。双酶切RAD虽然获得的片段数量少,但是不同样品在同一酶切位点处收集的片段长度一致,可以将read2部分也利用起来,利于后续设计探针等,更适合无参考序列的物种。想知道哪一种方法更适合自己的项目需要结合物种情况及研究目的综合得出。